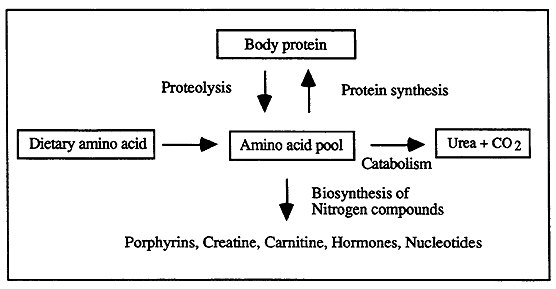
Protein Turnover / Ammonia Metabolism
Protein Metabolism and Nitrogen Economy:
A certain amount of dietary protein is required to synthesize endogenous proteins such as albumin (plasma protein), myosin (muscle filament), actin and hemoglobin. The basis for the dietary requirement of protein is the bodies inability to synthesize certain amino acids.
|
Essential
|
Non-Essential
|
|
Argininea
|
Alanine
|
|
Histidine
|
Aspartic
Acid
|
|
Isoleucine
|
Cysteine
|
|
Leucine
|
Glutamic
Acid
|
|
Lysine
|
Glycine
|
|
Methionineb
|
Proline
|
|
Phenylalaninec
|
Serine
|
|
Threonine
|
Tyrosine
|
|
Tryptophan
|
Glutamine
|
|
Valine
|
Asparagine
|
a: Arginine is synthesized by mammalian tissues, but the rate is not adequate during childhood.
b: Methionine is required in large amounts to produce cysteine if the latter is dietarily lacking.
c: Phenylalanine is needed in large amounts to produce tyrosine " ........................................".
Protein Turnover:
Protein Balance: interrelationship between protein synthesis and degradation (proteolysis).
Positive Nitrogen Balance: when dietary intake of proteins is greater than the requirement for endogenous protein synthesis.
Neutral Nitrogen Balance: dietary protein intake and endogenous proteins are maintained at a stable level (equilibrium).
Negative Nitrogen Balance: if protein intake is insufficient or if the balance of amino acids is incorrect for synthetic needs, endogenous protein is metabolized to liberate free amino acids for synthesis of essential proteins.
Nitrogen Economy:
Regulation of Protein Turnover and Nitrogen Economy:
Insulin: increases synthesis, decreases degradation ----> favors maintenance of body protein pool
Glucocorticoids: released during stress or starvation, oppose the effects of insulin and result in protein degradation (metabolism)
Insulin / Glucocorticoid Ratio
Fed State (+ Insulin): High ratio ----> Protein Synthesis
Fasting (- Insulin): Low ratio ----> Protein Degradation / Metabolism
Roles of Protein Degradation / Metabolism:
Digestion of dietary proteins
Activation of enzymes (zymogens)
Fuel supply
Maintain amino acid pools
Control organ growth
Tissue repair
Removal of abnormal proteins
Complement and blood clotting cascades
Some proteins degrade rapidly, t1/2 = minutes, others slowly, t1/2 = days.
Those that degrade rapidly may contain amino acid sequences which confer instability.
Others are "marked" by ubiquitin, a small protein forming a covalent linkage w/ proteins in an ATP dependent reaction.
Ammonia Metabolism and Nitrogen Waste:
The Transaminase Reaction:
Removes the a-nitrogen from the amino acid transferring it to the a-keto group of another organic acid, usually a-ketoglutarate.
Alanine + a-Ketoglutarate <----> Pyruvate + Glutamate
Aspartate + a-Ketoglutarate <----> OAA + Glutamate
Enzymes: amino acid specific, i.e. alanine transaminase, aspartate transaminase
Co-factor: pyridoxal phosphate, functions as a "Schiff" base intermediate
What happens to all of the Glutamate being produced? It is converted into a-KG and Ammonia (NH4+) by Glutamate Dehydrogenase
Glutamate + NADP+<----> a-Ketoglutarate + NADPH + NH4+
Glutamate DH: glutamate is transferred to the mitochondria where ammonia is liberated.
What happens to all of the NH4+?
Nitrogen Excretion:
The body removes nitrogen waste in a variety of forms.

The Urea Cycle:
Liver, minimal activity Kidney
Overall Reaction:
NH3 + CO2 + 3 ATP + Asp ----> Urea + Fumarate + 2 ATP + AMP + 2 Pi + PPi
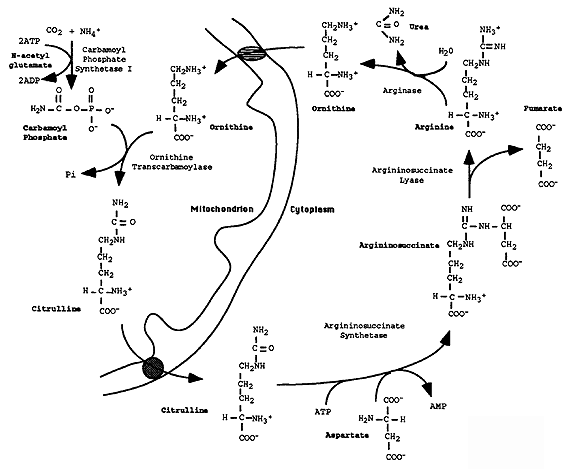
[1] Carbamoyl Phosphate Synthetase I (CPS I): provides the substrate, carbamoyl phosphate, for the urea cycle.
Allosteric Activation: N-Acetylglutamate via N-Acetylglutamate Synthase
Glutamate + Acetyl CoA ----> N-Acetylglutamate + CoA
[2] Ornithine Transcarbamoylase (OTC): citrulline is exported from the mitochondria to the cytosol in exchange for ornithine.
[3] Argininosuccinate Synthetase: uses two high energy phosphate bonds
[4] Argininosuccinate Lyase: removes all but the amino group from Asp, fumarate is recycled back to Asp via malate and OAA in mitochondria.
[5] Arginase: urea is produced and ornithine is regenerated.
Clinical Correlate:
Aquired Hyperammonemia: may result from cirrhosis of the liver and is only cured by a liver transplant
Inherited Hyperammonemia:
Cause: deficiencies of urea cycle enzymes, almost exclusively seen in children
Symptoms: severity depends on proximity of defect to point of entry of ammonia into the cycle (CPS I step) can be life-threatening to no symptoms at all. A total deficiency of an enzyme is usually fatal or results in severe mental retardation.
Causes of Neurologic Damage:
(1) ammonia reacts with a-ketoglutarate to form glutamate thus interfering with ATP production in the TCA cycle
(2) excess glutamate undergoes successive amination to glutamine then a-ketoglutaramic acid, a neorotoxic cmpd.
(3) The huge increases of a single amino acid in the blood result in limited availability of others within the brain, thus reducing rates of protein synthesis.
Treatment:
(1) Dietary restriction of proteins.
(2) Organic Acid Conjugation: Benzoic and Phenylacetic Acid
Benzoic Acid: conjugates with Gly and forms hippuric acid which is readily excreted in the urine. Gly can be synthesized from CO2 and NH3 in quantities needed for endogenous protein synthesis.
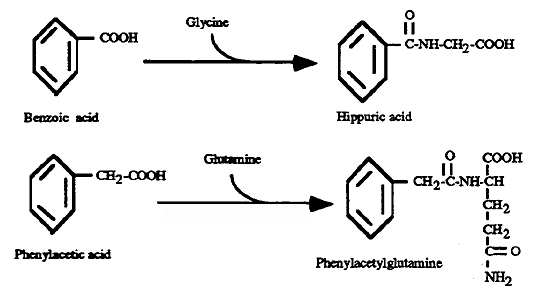
Phenylacetic Acid: conjugates with Gln forming phenylacetylglutamine which is readily excreted in the urine. Gln can also be synthesized in quantities needed for endogenous protein synthesis
.
© Dr. Noel Sturm 2015